Scheda malattia
Deficit di G6PD- Favismo
La carenza di glucosio 6 fosfato deidrogenasi (G6PD) è un difetto enzimatico ereditario la cui principale manifestazione clinica è costituita da una crisi emolitica acuta, talora gravissima, scatenata da ingestione di fave (favismo), infezioni o farmaci. Al di là del suo interesse cinico, questo disordine metabolico rappresenta un modello biologico che ha permesso importanti acquisizioni sulla regolazione della funzione genica nel cromosoma X e sulla sua struttura e di dimostrare l’origine clonale di alcune anomalie ematologiche
EPIDEMIOLOGIA
E stato calcolato che circa 100 milioni di persone nel mondo (tra maschi emizigoti e femmine etero-od omozigoti) sono portatori di almeno un gene per la carenza di G6PD. Nessuna parte del mondo è risparmiata, ma vi sono delle aree geografiche che presentano un’incidenza particolarmente elevata: Africa centro-settentrionale, bacino del Mediterraneo, Cina e India. Nell’Italia continentale l’incidenza media della carenza di G6PD è dello 0,4%, in Sicilia è dell’l% mentre in Sardegna raggiunge il valore medio di 14,3% con un picco del 25,8% nella provincia di Cagliari. Il fenomeno immigratorio degli ultimi decenni ha fatto si che anche i medici dell’Italia continentale possano osservare manifestazioni cliniche da carenza di G6PD.
L’elevata incidenza del gene per la carenza di G6PD nelle aree tropicali e sub tropicali caratterizzate, nel passato e ancor oggi, dall’endemia malarica ha fatto supporre che il difetto enzimatico esercitasse in qualche modo un effetto protettivo dall’infezione malarica. In effetti è stato dimostrato che nelle femmine eterozigoti lo sviluppo del Plasmodium falciparum è notevolmente ridotto nelle emazie carenti rispetto a quelle normali
BASI GENETICHE
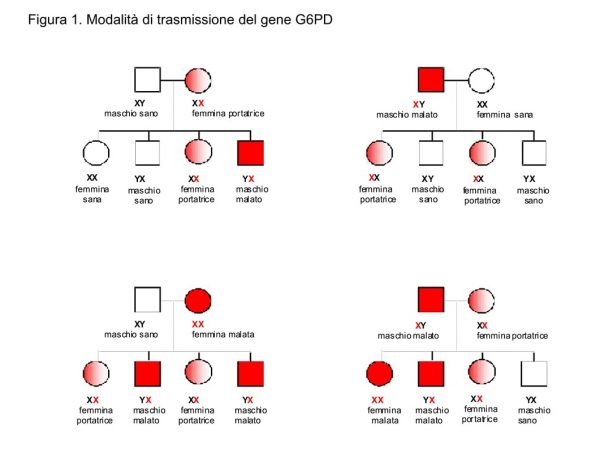
Il gene strutturale della G6PD si trova sul braccio lungo del cromosoma X (banda Xq28) e si trasmette secondo i meccanismi dell’eredità X linked (fig. 1)
Esistono più di 400 varianti di G6PD, distinte da caratteristiche biochimiche e funzionali le quali si associano ad una diversa suscettibilità agli stimoli ossidativi/emolitici. A seconda della variante molecolare del deficit e dell’attività residua dell’enzima nel globulo rosso è stata osservata una grande eterogeneità clinica, potendo variare da forme asintomatiche a forme gravi.
La maggior parte delle varianti sono rare, mentre sono molti frequenti la variante africana o GdA- e la variante mediterranea GdB-. A parte la diversa mobilità elettroforetica (aumentata nella prima, normale nella seconda), la differenza più importante tra le due varianti è costituita dalla differente velocità di degradazione dell’enzima.
L’attività dell’enzima normale ha un tempo di dimezzamento di 62 giorni, che si riduce a 8 giorni nella GdA-, ed è ancora minore nella variante mediterranea. Ne consegue che nella GdA- i globuli rossi appena immessi in circolo dal midollo (reticolociti) hanno un contenuto normale di enzima che però si riduce rapidamente con l’invecchiamento, mentre nella variante mediterranea il contenuto di enzima è molto ridotto già nei reticolociti e continua a ridursi con estrema rapidità. Per questa ragione l’attività enzimatica eritrocitaria determinata in laboratorio sarà fortemente ridotta rispetto al normale nella variante GdA- (5-15%) e praticamente assente nella variante mediterranea (0-5%).
Un’importante conseguenza pratica è la maggior gravità della crisi emolitica nella variante mediterranea rispetto alla GdA-. In quest’ultima, infatti, verranno distrutte solo le emazie più vecchie mentre i reticolociti appena immessi in circolo dal midollo come risposta all’emolisi verranno risparmiati per il loro contenuto enzimatico normale. Al contrario nella variante mediterranea sia le emazie circolanti che i reticolociti sono gravemente carenti, rendendo la crisi emolitica minacciosa per la vita.
La classificazione dell’OMS della carenza di G-6-PD si basa sul livello di attività eritrocitaria dell’enzima e sull’importanza delle manifestazioni cliniche:
- Classe I: deficit severo (dall’1 al 4 % di attività enzimatica residua).
- Classe II: deficit intermedio (dal 3 al 10 % di attività enzimatica residua).
- Classe III: deficit moderato (dal 10 al 40% di attività enzimatica residua).
PATOGENESI
La G6PD è un enzima citoplasmatico presente in tutte le cellule che catalizza la prima reazione della via dei pentoso-fosfati dalla quale si produce ribosio 5 fosfato, importante per la sintesi di nucleotidi ed il NADPH, coenzima e principale donatore di idrogeno nelle reazioni di biosintesi. Il NADPH è indispensabile per la riduzione del glutatione (GSH), tramite la glutatione-reduttasi. L’eritrocita utilizza il GSH per proteggere dalla denaturazione ossidativa i gruppi sulfidrilici (-SH) dell’emoglobina e della membrana cellulare. In assenza di GSH, l’emoglobina denaturata precipita all’interno della cellula formando i corpi di Heinz. La denaturazione dell’emoglobina e l’ossidazione dei costituenti di membrana sono responsabili dell’emolisi.
La G6PD è normalmente presente in tutti i tessuti ma il suo deficit si esprime essenzialmente nel globulo rosso in cui a differenza delle altre cellule nucleate dell’organismo nessun altro enzima permette la produzione di NADPH. In assenza di NADPH tutti gli stress ossidativi comportano un’alterazione dell’emoglobina e della membrana dell’eritrocita.
L’NADPH è inoltre responsabile della distruzione del perossido di idrogeno (H2O2), sostanza altamente tossica per la cellula.
QUADRO CLINICO
L’espressioni cliniche principali del deficit di G6PD sono
- Favismo: anemia emolitica acuta, indotta dall’assunzione di farmaci o alimenti (fave), o durante un’infezione
- Ittero neonatale, con sequele neurologiche nei casi più severi e non trattati
- Anemia emolitica cronica non sferocitica
Favismo
L’ingestione di fave è la più comune causa di crisi emolitica nei soggetti carenti di G6PD nella variante mediterranea, mentre non sembra provocare alcun disturbo negli individui con variante africana.
L’emolisi può verificarsi dopo il consumo di fave fresche, secche o surgelate (ma è più frequente nel primo caso) e anche in lattanti al seno le cui madri avevano mangiato fave. Tuttavia, sono noti casi di individui che avevano consumato fave in molte occasioni senza alcun disturbo prima di presentare una tipica crisi emolitica da fave; in altri casi, la temeraria assunzione dell’alimento anni dopo una crisi emolitica, non ha determinato disturbi. Pertanto è evidente che esistono altri fattori che contribuiscono allo scatenamento della crisi emolitica.
I soggetti più colpiti sono bambini maschi dei quali non era ancora nota la carenza enzimatica; naturalmente la stagione di maggior incidenza è la primavera quando si consumano le fave fresche. Alcune sostanze estratte dalle fave (divicina, isouramile) hanno una potente azione ossidante.
La crisi esordisce bruscamente, da poche ore a 1-3 giorni dopo l’ingestione delle fave. Il pallore rapidamente ingravescente, l’ittero sclerale, l’emissione di urine color rosso vino sono i segni principali. Possono esservi dolori addominali, ingrossamento della milza e febbre. Con il progredire dell’anemia (di solito ore) compaiono polipnea, tachicardia, ipotensione. La massiva emoglobinuria può determinare insufficienza renale acuta. Vi è reticolocitosi, leucocitosi con neutrofilia, probabilmente in seguito alla stimolazione midollare e spesso un moderato incremento delle transaminasi.
La morfologia eritrocitaria è assai caratteristica: emazie parzialmente prive di emoglobina o con la membrana interrotta, e frammenti di emazie. Queste alterazioni della membrana eritrocitaria sono probabilmente espressione del danno provocato dal reticolo endotelio splenico (‘”pitting”) nell’asportare i precipitati di emoglobina. Questi precipitati, corpi di Heinz, si possono evidenziare nello striscio di sangue periferico con opportune colorazioni.
Il sospetto clinico è facile per l’anamnesi (ingestione di fave, talora favismo nei maschi della famiglia materna), l’esordio brusco e i segni caratteristici, l’evoluzione progressiva e drammatica.
La diagnosi differenziale è con le forme di anemia emolitica autoimmune e con la sferocitosi.
Nelle forme a decorso meno grave dovrà porsi la diagnosi differenziale con emoglobinurie di altra natura (avvelenamenti, emoglobinuria parossistica notturna), con la mioglobinuria (dolori muscolari violenti, diverso spettro di assorbimento tra emoglobina e mioglobina), con l’epatite virale (non vi è anemia e l’ipocromia delle urirne è dovuta ai pigmenti biliari e non a emoglobina). L’anemia emolitico uremica (s. di Gasser), che pure presenta caratteristicamente frammentazione eritrocitaria (eritrociti a guscio d’uovo), potrà essere differenziata, oltre che per l’anamnesi, per la minore evidenza clinica dell’emolisi, la costante compromissione renale e la trombocitopenia.
Le femmine eterozigoti presentano di solito livelli di enzima eritrocitario intermedio fra il normale e l’omozigote maschio carente. Questo fenomeno si spiega con l’inattivazione funzionale di uno dei due cromosomi X di ciascuna cellula che si verifica in una fase molto precoce dello sviluppo embrionale. In conformità alla teoria di Lyon, esse sono in realtà mosaici, essendo alcuni globuli rossi provvisti dell’enzima normale ed altri invece della variante carente. Poiché l’inattivazione dell’uno piuttosto che dell’altro cromosoma X è casuale, le femmine eterozigoti hanno generalmente la metà del livello normale di enzima, ma può accadere che talune femmine abbiano livelli enzimatici normali o quasi (per inattivazione prevalente del cromosoma X con il gene anomalo) e altre livelli vicini allo zero (per inattivazione prevalente del cromosoma X con il gene normale).
La determinazione del livello di G6PD eritrocitaria nel paziente e nella madre confermerà la diagnosi. Naturalmente il dosaggio dell’enzima non può essere eseguito subito dopo una trasfusione, né immediatamente dopo una crisi emolitica, anche se non richiedente trasfusione, in quanto i globuli rossi giovani, appena immessi in circolo, sono più ricchi di enzima.
L’unico trattamento disponibile della crisi emolitica acuta è la trasfusione di emazie. Nelle aree ad elevata prevalenza del gene bisognerà aver cura di non trasfondere emazie carenti.
EMOLISI DA INFEZIONE E DA FARMACI
I soggetti carenti di G6PD possono andare incontro a emolisi di gravità molto variabile in corso di infezioni o in seguito ad assunzione di farmaci. E’ molto difficile dire con quale frequenza ciò avvenga perché il fenomeno emolitico non è, in questi casi, né obbligatorio né costante. A parte la possibilità di particolari varianti dell’enzima, altri fattori possono contribuire a determinare questa variabilità dell’effetto emolitico quali lo stato immunitario del paziente, la carica batterica o virale, la dose del farmaco e l’associazione di più farmaci. Inoltre, molto spesso ad una condizione infettiva, o presunta tale, si associa la prescrizione di farmaci (antibiotici e/o antipiretici), rendendo difficile l’attribuzione della responsabilità dell’emolisi. L’elenco dei farmaci capaci di causare emolisi è molto ricco, ma per molti di essi non esiste sicura dimostrazione che, alle dosi terapeutiche, possano essere nocivi. Poiché molti antipiretici sono sospettati di azione emolitica, nella pratica pediatrica si pone spesso il problema del controllo della febbre nei soggetti carenti. Il paracetamolo è stato utilizzato a dosi terapeutiche nei bambini carenti senza alcun problema emolitico.
Tra le infezioni associate a emolisi sono state segnalate salmonellosi, polmoniti, sepsi, epatite virale.
Per molti dei farmaci implicati l’azione emolitica si esplica attraverso la produzione di perossidi e di radicali liberi. Meno chiaro è il meccanismo attraverso cui le infezioni possono causare emolisi: è possibile che durante la fagocitosi i leucociti liberino fattori ossidanti che esplicherebbero la loro azione sui globuli rossi presenti.
ITTERO NEONATALE GRAVE
L’ittero neonatale rappresenta in ordine di frequenza la seconda manifestazione clinica da carenza di G6PD. In Sardegna circa il 30% dei neonati maschi carenti sviluppa l’ittero che richiede l’exsanguinotrasfusione. La carenza enzimatica non conduce obbligatoriamente all’ittero patologico neonatale, ma costituisce un fattore che aggrava la predisposizione del neonato all’iperbilirubinemia. Il quadro clinico non differisce da quello dell’ittero neonatale non immunologico, e i neonati carenti non mostrano una riduzione della massa eritrocitaria superiore a quella dei neonati sani, carenti e non. Pertanto, piuttosto che a un incremento della fisiologica emolisi dell’epoca neonatale, l’iperbilirubinemia dei neonati carenti può essere interpretata come espressione di un più accentuato deficit funzionale degli epatociti carenti.
La diagnosi differenziale in epoca neonatale si pone con le altre forme di ittero e in particolare con la Malattia Emolitica del Neonato da ABO o da Rh, con il deficit di piruvato chinasi e con la sferocitosi.
ANEMIA EMOLITICA CRONICA NON SFEROCITICA
Esistono delle varianti, molto rare, che si associano ad emolisi cronica, con espressione clinica di gravità variabile. E’ verosimile che tali varianti, denominate di classe I, determinino un difetto enzimatico così grave che le sue conseguenze si manifestano anche durante i normali eventi stressanti e non ci sia bisogno quindi di cause straordinarie, quale l’ingestione delle fave e/o di farmaci.
La presentazione clinica più frequente riguarda soggetti maschi, che di solito hanno avuto un ittero neonatale, che si è risolto e si ripresenta più avanti o che, talvolta, continua e si accompagna ad anemia. La milza è solitamente ingrandita. L’anemia è di solito normocromica o lievemente macrocitica, a causa della intensa reticolocitosi. La morfologia eritrocitaria non è particolarmente caratteristica e sono presenti i classici reperti associati con le anemie emolitiche.
In questi casi la diagnosi puo’ essere più difficile in quanto occorre dosare l’enzima, la cui attività deve risultare bassa e caratterizzare la variante per avere la conferma che si tratti di una variante di classe I. Bisogna escludere infatti la possibilità che il soggetto sia portatore di una variante di classe II o III, asintomatica nel periodo di stato e che l’anemia cronica non sferocitica dipenda da qualche altra causa (carenza di piruvato chinasi, anemia diseritropoietica, ellissocitosi che possono trovarsi in associazione con la carenza di G6PD).
La terapia non differisce da quella proposta per le altre forme di anemia emolitica cronica non sferocitica dovute a difetti degli enzimi della glicolisi. E’ importante, una volta posta la diagnosi, offrire il consiglio genetico alla famiglia: innanzitutto va accertato se la madre è eterozigote (in alternativa si potrebbe trattare di una mutazione de novo nel soggetto) e quindi con un rischio di ricorrenza del 50% per ogni successivo figlio maschio. In questo caso la variante deve essere caratterizzata in modo da offrire una diagnosi prenatale mediante l’analisi molecolare dei villi coriali; in alternativa, se la variante non è stata identificata, una dimostrazione indiretta si puo’ ottenere mediante lo studio dei polimorfismi dei geni vicini a quello della G6PD nella madre e nel feto.
INDAGINI DIAGNOSTICHE
Nel deficit di G6PD, in crisi emolitica, è presente anemia normocromica-normocitica, reticolocitosi, e iperbilirubinemia indiretta; lo striscio di sangue periferico frammenti eritrocitari ed i corpi di Heinz. Il DAT (direct agglutination test o test di Coombs) è negativo. Il dosaggio dell’enzima è generalmente ridotto. Durante la crisi la reticolocitosi può portare a un dosaggio ai limiti della normalità del G6PD, anche se nei soggetti omo/emizigoti, anche in presenza di un elevato numero di reticolociti, il dosaggio enzimatico permette di fatto la diagnosi. Solo nelle femmine eterozigoti con basso numero di eritrociti G6PD carenti la determinazione spettrofotometrica dà valori sovrapponibili alla norma. In questo caso sarà utile il test citochimico di Sansone o il dosaggio della 2-deossi-glucosio-6-fosfato.
In alcune regioni è stato attivato lo screening neonatale per G6PD, come parte dello screening metabolico allargato.
INDAGINI DIAGNOSTICHE
Il Deficit di G6PD (Glucosio-6-fosfato-deidrogenasi), anche conosciuto come “Favismo”, è la carenza enzimatica più comune.
Il difetto enzimatico può provocare crisi emolitiche, cioè una distruzione dei globuli rossi, quando il soggetto carente ingerisce alcuni farmaci ossidanti o alimenti come le fave (da cui deriva il nome). Caratteristica dei fabici è di essere asintomatici fino a quando non si entra in contatto con i sopramenzionati agenti ossidanti. Si riporta un elenco sintetico dei più comuni farmaci da evitare. Per un elenco completo, consultare il documento a cura dell’Istituto Superiore di sanità: http://www.iss.it/binary/publ/cont/0947web.pdf
Alimenti da evitare
Fave
Attenzione ai preparati di ortaggi misti che possono contenere fave, tipo minestroni freschi o surgelati
Farmaci da evitare:
Antibiotici e chemioterapici:
Cloramfenicolo (Antibioptal, Betabioptal, Chloromycetin); Ciprofloxacina (Basemar, Battizer, Cexidal, Ciperus, Generflon, Mediflox); Ofloxacina, Levofloxacina, Trovafloxacina, Moxifloxacina, Norfloxacina, Pefloxacina, Rufloxacina; Acido nalidixico (Betaxina, Nalidixim, Naligram, Nalissina, Uralgin, Uriflor, Urogram, Uropan); Furazolidone (Furazone, Ginecofuran, Tricofur); Nitrofurantoina (Cistofuran, Furadantin, Furedan, Furil, Nitrofur, Urolisa, Acrodantin, Neofuradantin); Rasburicase (Fasturtec); Sulfacetamide (Minims Sulfacet, Prontamid, Sulfacetamide sod, Brumeton, Visublefarite, Aureomix, Chemyterral, Cosmiciclina); Sulfametoxazolo (Gantanol, Abacin, Bacterial, Bactrim, Chemitrim, Eusaprim, Gantrim, Isotrim, Medixin, Streptoplus, Suprin valeas).
Analgesici e antipiretici:
Acido acetilsalicilico; Fenazopiridina (Pyridium).
Antielmintici:
Niridazolo (Ambilhar)
Antimalarici:
Chinina; Primachina (Primaquine, Primachina, Plasmachina, Clorochina).
Chemioterapici:
Doxorubicina (Adriblastina); Sulfanilamide (Exoseptoplix, Solfanilamide, Otocaina); Sulfapiridina (Salazopyrin en, Salisulf, Lagena).
Blu di metilene (Terapia metaemoglobinemia; Evidenziatore fistole; In pazienti con metaemoglobinemia) e Blu di toluidina (Evidenziatore tumori).
FARMACI CONSIDERATI SICURI
In caso di necessità possono essere assunti senza pericolo di crisi emolitica: ampicillina, amoxicillina, amoxicillina + ac. clavulanico, cefalosporine, macrolidi, ureidopenicilline, aminoglicosidi, aminopeptidi.
In caso di necessità può essere assunto senza pericolo di crisi emolitica: paracetamolo